



AI and machine learning are conquering the world of medical devices. These innovative technologies are not only being used in health software such as SaMD, but are also increasingly finding their way into devices and systems as embedded software. While the FDA has already approved a number of AI/ML-based medical devices in the USA, Europe is somewhat more hesitant. But what do manufacturers in this country actually have to consider if they want to integrate these state-of-the-art achievements into their products? Our article sheds some light on this: it deals with the expansion of the QM system and its processes, additional activities in the software life cycle processes as well as in clinical evaluation and usability assessment.
Regulatory requirements for AI-/ML-based medical devices
At the beginning of 2021, the US FDA published an “Artificial Intelligence and Machine Learning (AI/ML) Software as a Medical Device Action Plan“, the Notified Bodies published version 5 of their “Questionnaire “Artificial Intelligence (AI) in medical devices” at the end of 2023 and the AI ACT was adopted in the EU on 21 May 2024 and must now be implemented into national law.
In addition to these regulations, there are already several standards on artificial intelligence, most of which are not specific to medical technology:
- ISO/TR 24291:2021 – Health informatics, Applications of machine learning technologies in imaging and other medical applications
- ISO/IEC 23894:2023 – Information technology – Artificial intelligence, Guidance on risk management
- ISO/IEC TR 24027:2021 – Information technology – Artificial intelligence (AI), Bias in AI systems and AI aided decision making
- ISO/IEC TR 24028:2020 – Information technology – Artificial intelligence, Overview of trustworthiness in artificial intelligence
- ISO/IEC TR 29119-11:2020 – Software and systems engineering – Software testing, Part 11: Guidelines on the testing of AI-based systems
- ISO/IEC TR 24029-1:2021 – Artificial Intelligence (AI) – Assessment of the robustness of neural networks, Part 1: Overview
- ISO/IEC 24029-2:2023 – Artificial intelligence (AI) – Assessment of the robustness of neural networks, Part 2: Methodology for the use of formal methods
- ISO/IEC 8183:2023 – Information technology – Artificial intelligence, Data life cycle framework
- IEEE 2801-2022 – IEEE Recommended Practice for the Quality Management of Datasets for Medical Artificial Intelligence
In the near future, the authorisation procedures and regulatory requirements for processes and medical devices in the EU and US markets will therefore be largely concretised. As a European manufacturer, it is worth taking an early look at the AI Act.
What are the necessary adjustments to the QM system and its processes?
Medical device manufacturers face a particular challenge in order to always fulfil the state of the art required for approval. This is because the underlying technologies are constantly evolving, while the regulatory requirements often lag behind.
As a result, the process for updating regulatory requirements must become much more dynamic – from an annual cycle to at least a quarterly cycle. The following data sources, among others, must be taken into account to determine the current state of the art:
- European Commission website on the progress of European AI legislation
- Website of the FDA, Health Canada, the MHRA (UK) and other national authorities on their AI legislation (as these are the “key opinion leaders”)
- Websites of the standards organisations ISO, IEC, IEEE on standards and draft standards
- Website of the Interest Group of Notified Bodies (IG-NB) on their positioning
- Websites/tools of the manufacturers of AI/ML technology, e.g. Google AI
The following processes, among others, must be expanded in the quality management system:
- Software life cycle processes
- Risk management
- Usability Engineering
- Data management
- Verification & validation
- Clinical evaluation
- User information/communication via the user interface and the IFU
- Installation, training, maintenance and service processes
- Post Market Surveillance.
What about extending software lifecycle processes?
Data management plays a key role in the quality and therefore the success of medical devices with AI and machine learning. This involves the data used for training, evaluation/validation and subsequent optimisation of the AI model. It should be noted that the EU AI law categorises medical devices as high-risk AI systems.
Specifically, Article 10 of the law requires:
- (2) Appropriate data governance and data management practices shall be subject to training, validation and testing data sets. […]
- (3) The training, validation and testing data sets shall be relevant, representative, free of errors and complete. They shall have the appropriate statistical properties, including, where applicable, as regards the persons or groups of persons to which the high-risk AI system is intended to be used. […]
To implement these requirements, medical technology companies generally need experts in data management first – a resource that has not been available in many companies to date.
In addition, the software development process in accordance with EN 62304 must be expanded to include specific activities relating to the AI model, as shown in the following chart:
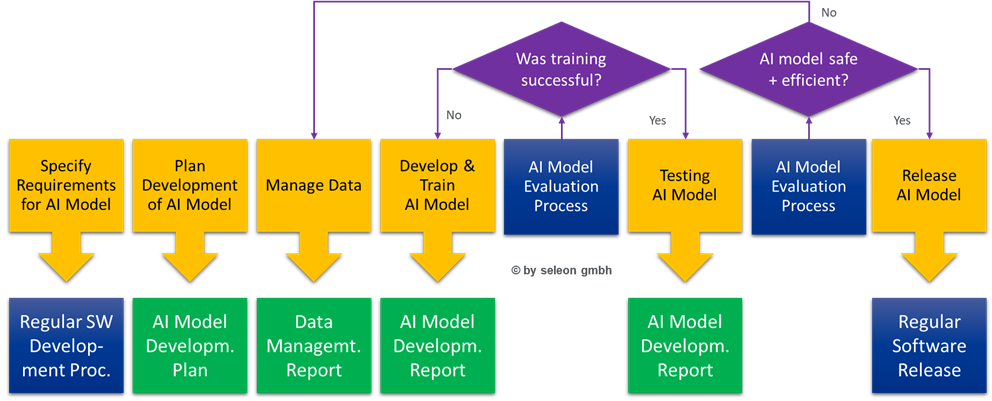
Illustration 1 Additional AI model-related activities in the software development process
This is the only way manufacturers can fulfil the regulatory requirements for the use of AI and machine learning in medical devices.
The software maintenance process also needs to be expanded accordingly in order to support the continuous further development of the AI model in line with regulatory requirements.
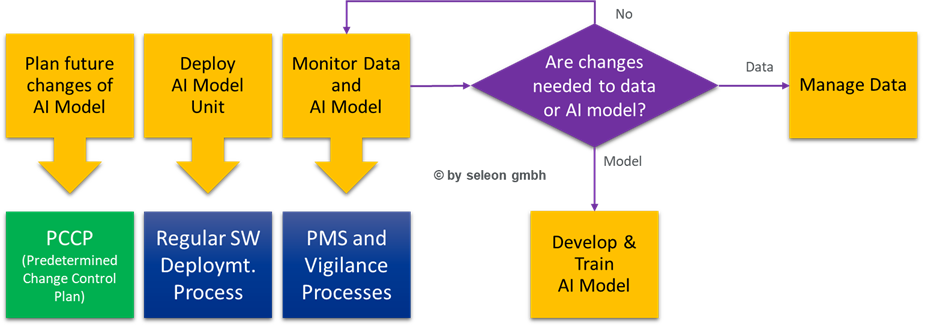
Illustration 2 Additional AI model-related activities in the software maintenance process
Is it also necessary to extend usability engineering?
The rather implicit usability requirements of the Medical Device Regulation (MDR) must be adapted accordingly, particularly with regard to the behaviour of the AI model in the medical device, which is often intransparent to the user.
This includes the following specific usability requirements for medical devices with AI and machine learning:
Usage environment usability tests: Usability tests must be conducted in an environment that adequately reflects the actual clinical work environment and the cognitive load of the user.
- Workflow management: The clinical workflow requirements must be identified and translated into the AI-based software specifications for the medical device.
- User group and patient population: The requirements for user input and the target patient population must be specified, taking into account the level of training of the users.
- Transparency: The system outputs must be transparent for the user and correspond to their level of understanding.
- Explainability: The data processing and its meaning must be explainable to the user and consider their level of training.
- Automated biases: The presence of automation bias must be analysed.
- Error handling: Error messages must be transparent and understandable for the user. It must also be possible to hand over the system control to the user if the system is unable to fulfil its purpose.
- Update management: Information on system updates (model or data) must be presented to the user in a way that is easy to understand in terms of type, reason and impact on performance and security.
Practical assistance for the implementation of these usability requirements can also be found in the “Good Machine Learning Practice for Medical Device Development: Guiding Principles” by the US FDA, the Canadian Health Canada and the British MHRA:
- Point 1 calls for multidisciplinary expertise throughout the product lifecycle:
An in-depth understanding of a model’s intended integration into the clinical workflow, and the desired benefits and associated patient risks, can help ensure that ML-enabled medical devices are safe and effective and address clinically meaningful needs over the lifecycle of the device. - Point 7 emphasises the performance of the human-AI team:
Human factors considerations and human interpretability of the model outputs are best addressed when the model has a “human in the loop”, where the emphasis should be more on the performance of the human-AI team than on the performance of the model on its own. - Point 9 addresses user information:
Users are provided with ready access to clear, contextually relevant information that is appropriate for the intended audience (e.g. healthcare providers or patients). This includes the intended use and indications for use of the device, the performance of the model for appropriate subgroups, the characteristics of the data used to train and test the model, acceptable inputs, known limitations, user interface interpretation, and how to integrate the model into the clinical workflow. Users are also made aware of device modifications and updates from real-world performance monitoring, the basis for decision making and the means to communicate concerns about the product to the developer
Expanding the usability engineering process to include AI model-related activities would look like this:
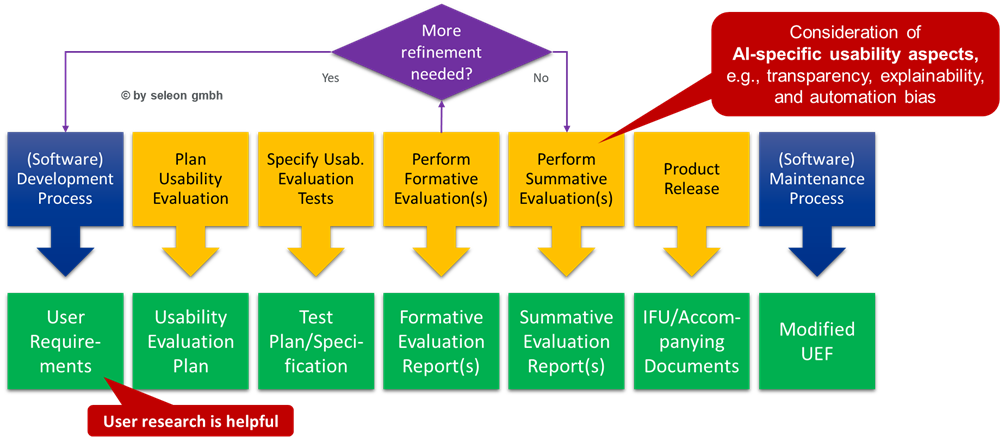
Illustration 3 Additional AI model-related activities in usability engineering
How is this implemented in the clinical evaluation?
In the clinical evaluation, the manufacturer must (be able to) provide evidence (at any time),
- that the medical device provides the intended performance,
- is suitable for its intended purpose,
- is safe and effective,
- does not jeopardise the clinical condition or the safety/health of patients, users or other persons and
- has a positive benefit-risk ratio.
Of course, this also applies to medical devices with AI/ML technology, where the following specific requirements must be taken into account:
- The clinical evaluation plan must identify the state of the art in science and technology in the relevant medical field:
- The technical SOTA in relation to AI must be identified
- Proof of clinical relevance and scientific validity must be provided
- The plan for the data query must address a suitable database search:
- Literature search in PubMed and ClinicalTrials.gov with specific medical terms (medical subject headings, MeSH), such as “Machine Learning”.
- The assessment of the clinical data must be carried out taking appropriate criteria into account:
- Analysing the quality of literature data on clinical trials using the CONSORT-AI and SPIRIT-AI1 criteria
- Analysing the quality of literature data related to diagnostic accuracy studies based on the STARD 2015 criteria
- Independence of the test data set from the training data set (or information on data splitting) in studies on diagnostic accuracy
- Use of an external test dataset in addition to an internal test dataset in diagnostic accuracy studies to test the generalisability of the model
- Appropriateness and reproducibly correct calculation of quality measures in clinical trials and diagnostic accuracy studies
- The clinical evaluation report must document the requirements for safety and an acceptable benefit/risk profile as well as the requirements for performance:
- Proof of the intended medical benefit at the specified values of the defined quality measures
- Comparison of the product to be evaluated with conventional clinical diagnostic or treatment procedures (reference standard)
- Proof of technical/analytical capability
- Prospective, randomised, multicentre, state of the art study to confirm generalisability and investigate use beyond the intended purpose
- Proof of clinical performance in terms of discrimination performance, calibration performance and clinical acceptability
So what needs to be considered from a regulatory perspective for medical devices with AI/ML technology?
We summarise the most important points from this article as follows:
- Conformity assessment procedures are carried out in the EU in accordance with the MDR, IVDR and later the EU AI Act; in the USA, they are carried out via the established authorisation procedures, with technology-specific features
- The state of the art is constantly changing, which increases complexity.
- The quality management system and its processes must be expanded for additional AI/ML topics and requirements.
- Data processing issues are very important, especially the independence of the test data from the training data. Qualified data managers are required.
- The software life cycle processes must be expanded to include additional activities and further documents must be created.
- Usability engineering must take into account AI/ML-specific aspects such as transparency, explainability and automated bias.
- Special requirements for workflow management, patient population and user group must be met.
- The clinical evaluation must take into account the AI/ML-specific state of the art and the relevant literature.
- Prospective, randomised, multi-centre, state-of-the-art studies are usually a prerequisite for successful clinical evaluation of medical devices with AI/ML technology to confirm generalisability and use beyond the intended purpose.
- Static AI can be approved in the “classic way”, dynamic AI is currently not certifiable in principle and static black box AI can be approved by a case-by-case decision of the Notified Body.
Are you taking a chance to use AI/ML technology in your medical device? We will be happy to help you build up your knowledge and the knowledge of your product and support you throughout the entire life cycle.
Please note that all details and listings do not claim to be complete, are without guarantee and are for information purposes only.

