



For the treatment of some diseases, it may be necessary to combine medical devices with medicinal products. This quickly results in a so-called “combination product” – Drug Device Combination. But what requirements apply to medical devices that are combined with a medicinal product? In this article we show important background information and correlations that are essential in order to fulfil the regulatory requirements, which have definitely become more complex with the entry into force of the MDR.
“Combination products” and how they are defined
First, it is worth noting that the term “combination product” per se is not used in the MDR. Rather, in the defining Article 1, paragraphs 8 and 9 speak of a single integral product or integral part of a product. The principal intended action of the product is decisive for the definition. Thus, the question must first be answered as to which of the two components provides the principal intended action and which intended purpose specifically results from this. These two factors determine whether the product is primarily classified as a medical device or as a medicinal product. If the principal intended action can be clearly attributed to a pharmacological, immunological or metabolic mode of action, the product is regulated under the legislation for medicinal products (primarily the respective Medicinal Products Act – Arzneimittelgesetz AMG in Germany – and Directive 2001/83/EC). This is also referred to as a medical device with medicinal products as an integral part.
If, however, the principal intended action emanates from the medical device and the integral, pharmaceutical component has only a supporting function, the entire product is treated as a medical device.
In case of doubt, the MDR and the Medicinal Products Act should be intensively evaluated or a request for clarification should be sent to the BfArM or the respective local competent authority.
It is important to note that products for dispensing medicinal products, systems and treatment units are not combination products. Depending on whether the product is primarily classified as a medicinal product or a medical device, the respective other authority must still be involved.
To help you classify a possible “combination product”, please find here our flowchart on this topic:
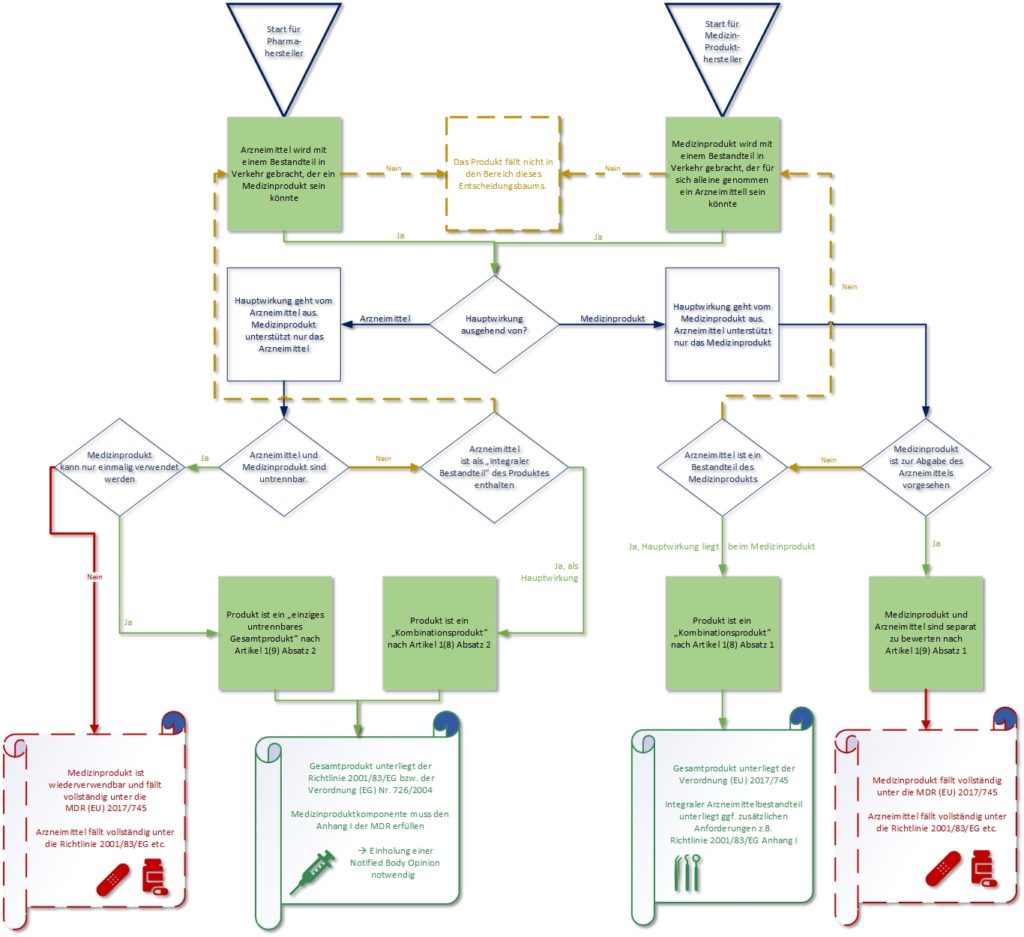
When is the product applicable for the scope of Directive 2001/83/EC?
In determining which scope is applicable for the product in question, the principal intended action is decisive. Any product that contains as an integral part a substance that would be considered a medicinal product on its own, but which has only an auxiliary function in the product-related application, is assessed and authorised on the basis of the MDR.
However, if this substance is assigned a principal function in the mode of action of the overall device, Directive 2001/83/EC on medicinal products applies to the entire device. In this case, the relevant general safety and performance requirements listed in Annex I of the MDR apply to the safety and performance of the medical device part (MDR, Article 1 (8)). Any product intended to deliver a medicinal product (as defined in Directive 2001/83/EC) falls within the scope of the MDR, the provisions from the Medicines Directive do not then apply.
However, if the device intended to deliver a medicinal product and the medicinal product are placed on the market in such a way that they form a single integral product intended exclusively for use in that combination and not being reusable, that single integral product shall be subject to the Directive 2001/83/EC or Regulation (EC) No 726/2004. In that case, the safety and performance of the medical device part in that product shall be subject to the general safety and performance requirements set out in Annex I to the MDR. (MDR, Article 1 (9))
In addition, Article 117 of the MDR / Annex I Section 3.2 point 12 of Directive 2001/83/EC requires manufacturers who place a combination of medicinal product and medical device on the market as a device with an integral part and market it as a “medicinal product” to obtain an opinion from a Notified Body, the so-called Notified Body Opinion. The Notified Body then confirms that the device complies with the general safety and performance requirements of MDR (EU) 2017/745 and sends an opinion to the competent medicine authority confirming this compliance. A certificate is issued to the manufacturer, which becomes part of the application for marketing authorisation. This applies in particular to new marketing authorisations or substantial changes to the products. There is a Team-NB position paper on this topic, which provides guidance on how to prepare and submit the relevant documents.
Products that may fall into this category include auto-injectors, inhalers, pre-filled nebulisers or syringes.
A brief excursion into pharmaceutical law
When dealing with “combination products”, it makes sense to delve a little deeper into the law on medicinal products and also to define the term “medicinal product” more closely. In the associated Directive 2001/83/EC, the term “medicinal product” is defined in Article 1 (2) as follows:
Any substance or combination of substances
- presented for treating or preventing disease in human beings;
- which may be administered to human beings with a view to making a medical diagnosis or to restoring, correcting or modifying physiological functions in human beings
are considered medicinal products.
In the production and processing of the individual substances, it is important to follow and act in accordance with the GMP guidelines, which are regulated in Germany by the Ordinance on the Manufacture of Medicinal Products and Active Pharmaceutical Ingredients (AMWHV).
The European Medicine Agency is the authority that is mainly involved in the regulation of medicinal products. In the context of combined products, the EMA can therefore also become relevant.
Challenges in ensuring product safety
Already in the regular conformity assessment of medical devices, biological evaluation and biocompatibility play a significant role. However, in combination with medicinal products, this becomes even more important..
For example, in the biological evaluation and material characterisation, the focus can be on substances such as phthalates, SVHC substances, CMR substances or other biological substances. In order to be able to guarantee absolute transparency, critical components must be sampled and evaluated; the list of possible choices here is very long and becomes even more complex when combined with a medicinal product. The so-called APIs – active pharmaceutical ingredients – also play a role here and must also be characterised and tested. The difference between active ingredient and excipient in the formulation is also decisive here and makes the procedure more complex.
Since the principal intended action of the overall product takes place in two different ways and thus there are more sources of risk, the mechanism of biological evaluation counteracts this and places particular emphasis on product safety.
For all products that have to comply with Annex I of the MDR, it is also necessary to assess interactions and other aspects of product safety mentioned in Annex I of Directive 2001/83/EC. This requirement results from GSPR 12.2 of the MDR and highlights the points of absorption, distribution, metabolism, excretion, local tolerance, toxicity, interactions with other medical devices, medicinal products or other substances, and possible adverse reactions. For all “combination products” according to Article 1(8)(1), the aspects of quality, safety and usefulness of the substance according to Annex I of Directive 2001/83/EC must also be reviewed.
Technical documentation for the Notified Body Opinion
Manufacturers who need to obtain a Notified Body Opinion in order to provide objective evidence of compliance with Annex I should not solely focus on Annex I of the MDR. The evidence is provided in accordance with the modalities of Annex II Section 4. And the evidence itself can also be compiled in total on the basis of the requirements of Annex II “Technical Documentation” – this also applies with regard to the verification and validation of products and processes. The requirements for the UDI and the registration of economic operators certainly deviate from this. As far as the contents of the dossier are concerned, however, it helps to take Annex II into account. This is also addressed in the already mentioned Team-NB Position Paper “Documentation Requirements for Drug Device Combination Products”. The requirements of Annex III “Technical documentation on post-market surveillance” are already extensively covered by pharmacovigilance.
Instructions for the first steps
Identifying which requirements need to be considered and implemented for your product is not always easy. However, there are guidance documents, such as the MDCG‘s “on borderline between medical devices and medicinal products”, which is a very good guide for the first steps and should definitely be consulted.
However, if you prefer a personal touch, feel free to contact seleon’s experts, who can provide competent advice directly suited to your individual situation.
Please note that all details and listings do not claim to be complete, are without guarantee and are for information purposes only.

