
Ob kabellose Fußschalter, mobile Monitore oder tragbare Analysegeräte: Wo Batterien drinstecken, greift die Batterieverordnung (EU) 2023/1542. Der August 2025 war für alle Marktteilnehmer, die von der Batterieverordnung betroffen sind, ein spannender Monat mit neuen Pflichten. Aber aufgepasst: Mit dem Omnibus Paket IV wurden Pflichten verschoben. Was müssen Medizinproduktehersteller, die Batterien in Ihre Geräte verbauen, nun beachten? Wir klären auf über Anwendungsbereiche, Pflichten und Termine.
Wofür eine Batterieverordnung?
Batterien sind aus unserem klinischen Alltag nicht mehr wegzudenken – enthalten in kabellosen Fußschaltern bis hin zu mobilen Monitoren oder smarten Datensammlern. Doch was passiert eigentlich mit den Kraftpaketen, wenn ihr Leben zu Ende geht? Genau hier setzt die Batterieverordnung der EU an, mit dem Ziel den gesamten Lebenszyklus von Batterien umweltgerecht, sozialverträglich und sicher zu gestalten. Damit reiht sich die Verordnung in eine Reihe von Umweltgesetzgesetzgebungen der EU ein und erweitert gleichzeitig das Aufgabenfeld Ihrer Product Compliance.
- Am Juli 2023 wurde die Verordnung (EU) 2023/1542 vom Europäischen Parlament und dem Rat verabschiedet.
- Sie ist seit dem 18. Februar 2024 in weiten Teilen anwendbar. Ab dem 18. August 2025 ersetzt sie vollständig die bisherige Batterierichtlinie 2006/66/EG.
- Als Verordnung gilt sie unmittelbar in allen EU-Mitgliedstaaten.
- Viele Artikel werden durch delegierte Rechtsakte ergänzt.
- Anwendungsfristen für Sorgfaltspflichten des Artikel 48 wurden durch den Omnibus (EU) 2025/1561
Wen betrifft die Batterieverordnung – Erklärung von Rollen
Die Verordnung nimmt alle wirtschaftlichen Akteure entlang der Lieferkette in die Pflicht – vom Batteriehersteller bis zum Händler. Für die Medizintechnik sind diese Rollen von Relevanz:
- Erzeuger
- Einführer
- Händler
- Hersteller von Geräten mit Batterien
- Bevollmächtigter Vertreter
Wann gilt der Medizinproduktehersteller als Erzeuger einer Batterie, wann als Einführer oder Händler? Je nach Rollenzuweisung, kommen verschiedene Pflichten auf den Medizinproduktehersteller zu. Zur Veranschaulichung haben wir von seleon vier Szenarien definiert und im untenstehenden Schaubild dargestellt.
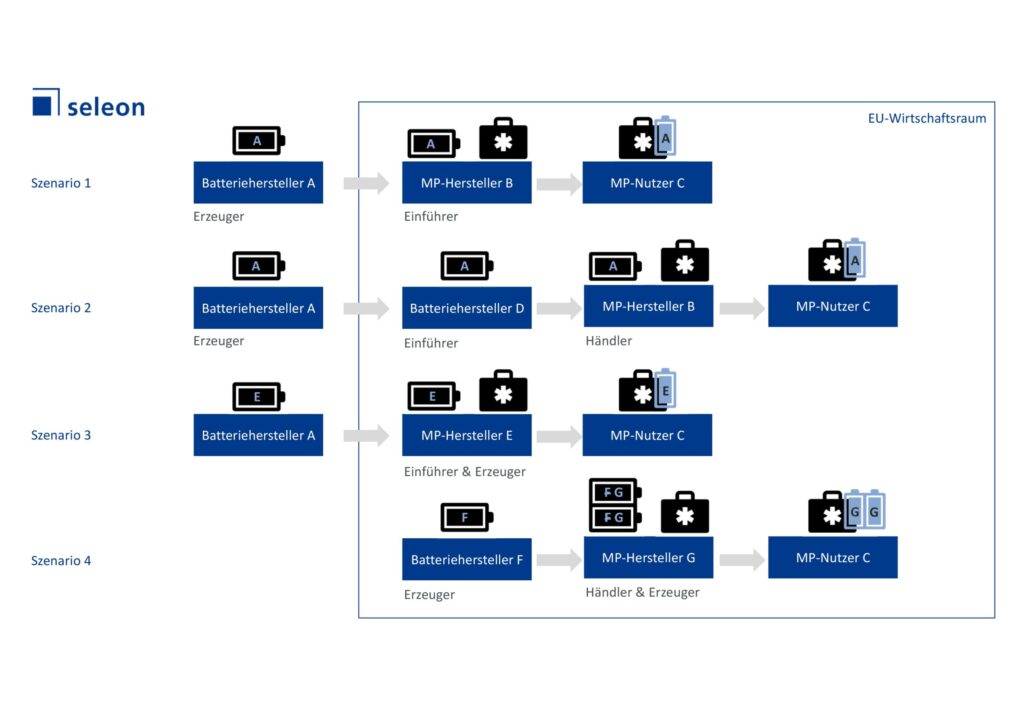
Name Bild: Rollen von Medizinprodukteherstellern (MP-Hersteller) in der Batterieverordnung
Was Sie sich fragen sollten: Sind Sie erstmaliger Inverkehrbringer im EU-Wirtschaftsraum (EWR)? In den meisten Fällen wird der MP-Hersteller eine Batterie kaufen, welche sich bereits im EWR in Verkehr befindet, und übernimmt somit die Rolle des Händlers (Szenario 2). Führt er die Batterie von einem Erzeuger aus einem Drittland ein, wird er zum Einführer (Szenario 1). Der MP-Hersteller kann jedoch nach Artikel 44 auch die Rolle des Erzeugers zugewiesen bekommen, und zwar wenn:
- er eine Batterie unter seinem eigenen Namen oder seiner eigenen Handelsmarke in Verkehr bringt oder sie in Betrieb nimmt (Szenario 3),
- er eine bereits in Verkehr gebrachte oder in Betrieb genommene Batterie so verändert, dass die Konformität mit den einschlägigen Anforderungen der Verordnung beeinträchtigt werden könnte (Szenario 4), oder
- er den Verwendungszweck einer bereits in Verkehr gebrachten oder in Betrieb genommenen Batterie verändert.
Fünf Batteriekategorien – die Gerätebatterie im Fokus
Weiterhin wird zwischen fünf Batteriearten (Gerätebatterien, Industriebatterien, Elektrofahrzeugbatterien, LV-Batterien, Starterbatterien) unterschieden. Für die Medizintechnik ist vor allem die Kategorie der Gerätebatterien entscheidend. Dabei ist es für die Definition unwesentlich, ob die Batterien fest oder austauschbar verbaut, wiederaufladbar oder für die Einwegnutzung ausgelegt sind. Solange die Batterie
- gekapselt ist,
- 5 kg oder weniger wiegt und
- keine Industriebatterie, Elektrofahrzeugbatterie, eine LV-Batterie oder eine Starterbatterie ist,
gilt sie als Gerätebatterie. Gerätebatterien sind wiederum teilweise als „Allzweck-Gerätebatterien“ kategorisiert – also Batterien wie wir sie auch im täglichen Haushalt nutzen und speziell auf Interoperabilität ausgelegt sind. Nachfolgend erläutern wir ausschließlich Pflichten, welche für (Allzweck-) Gerätebatterien gelten, da diese im Hauptfokus der Medizintechnik stehen.
Was für Pflichten kommen auf Medizinproduktehersteller bzgl. Gerätebatterien zu?
Sie sind Hersteller von Medizinprodukten oder In-vitro-Diagnostika und bauen Gerätebatterien nach der obigen Definition in ihr Produkt ein? Dann kommen einige Pflichten auf Sie zu – eine Übersicht findet sich in der untenstehenden Tabelle. Wichtig ist vorab zu klären, welche Rolle Sie bei dem Einbau von Gerätebatterien in Ihre Produkte einnehmen (siehe obiges Kapitel). Je nach Rolle, kommen verschiedene Pflichten auf Sie zu. 
Bildunterschrift: Pflichten für Gerätehersteller mit integrierten Batterien
Als Erzeuger sind Sie verpflichtet, die genannten Artikel 6-20 einzuhalten. Sind Sie Einführer oder Händler, müssen Sie die Einhaltung der Pflichten durch den Erzeuger garantieren. Hierunter fallen Pflichten zur Stoffbeschränkung, Leistung / Haltbarkeit, Kennzeichnung und Konformitätsbewertung. Alle Marktteilnehmer in der EU müssen eine Marktüberwachung zu ihren Produkten einführen und bei Bedarf Korrekturmaßnahmen zur Einhaltung der Pflichten einleiten.
Etwas mehr Zeit zum Durchatmen gibt es durch den Omnibus für Sorgfaltspflichten entlang der Lieferkette: Der Anwendungsbeginn wurde um zwei Jahre verschoben. Zeit, damit die Inverkehrbringer oder Betreiber der Batterien Strategien, Kontrollmechanismen sowie Risikobewertungen zur Sorgfaltspflicht in ihre Managementsysteme integrieren und durch benannte Stellen prüfen lassen können. Der Knackpunkt: Vielerorts gibt es noch keine zertifizierten Stellen, denn auch hier wurde der Zeitrahmen der Behörden um ein Jahr auf August 2026 verschoben.
Hinzu kommen je nach Rolle erweiterte Herstellerverantwortungen zum Sammeln von Altbatterien sowie der Finanzierung des Systems. Selbst wenn Sie nicht der Inverkehrbringer der Batterie sind, sondern lediglich mit diesen handeln: Sie sind zur Rücknahme von Altbatterien verpflichtet. Benannte Vertreter können hier jedoch Pflichten übernehmen.
Ob Erzeuger, Einführer oder Händler: Der Artikel 11 „Entfernbarkeit und Austauschbarkeit von Gerätebatterien“ ist für alle Medizinproduktehersteller anwendbar. Was müssen Sie also nun beim Design der Medizinprodukte beachten?
Pflicht zur Entfernbarkeit der Batterie durch Nutzer auch für Medizinprodukte?
Gerätebatterien müssen grundsätzlich ab dem 18.02.2027 durch Endnutzer entfernbar und austauschbar sein – ohne Spezialwerkzeuge, Wärme, Lösungsmittel oder herstellerspezifische Tools. Für Hersteller von Medizinprodukten und In-vitro-Diagnostika können jedoch einige Ausnahmen gelten. Denn professionelle medizinische Bildgebungs- und Strahlentherapiegeräte sowie In-vitro-Diagnostika sind von dieser Pflicht ausgenommen. Voraussetzung: Ein Austausch der Gerätebatterie durch unabhängige Fachleute ist möglich.

Bildunterschrift: Pflichten für Gerätehersteller mit integrierten Batterien
Auch wenn Sie nicht unter diese Ausnahme fallen, kann von weiteren Ausnahmen zu Art. 11 Gebrauch gemacht werden, wenn
- die Geräte in einer feuchten Umgebung eingesetzt werden,
- die Kontinuität der Stromversorgung notwendig ist und eine dauerhafte Verbindung zwischen Batterie und Produkt erforderlich ist, um Nutzer- oder Gerätesicherheit zu gewährleisten oder
- die Hauptfunktion des Produkts das Sammeln und Liefern von Daten ist und eine Unterbrechung der Stromversorgung die Datenintegrität gefährdet.
Wenn eine Ausnahme der Pflicht Anwendung finden soll, so ist dies anhand einer technischen Bewertung zu kennzeichnen und in der technischen Dokumentation zu dokumentieren. Die Leitlinie C/2025/214 (Anhang I, Abschnitt 5) gibt hierzu detaillierte Bestimmungen.
Was gilt es als Hersteller nun zu tun?
Sie sind Hersteller eines Medizinprodukts mit integrierter Batterie? Folgendes gilt es zu prüfen: Welche regulatorische Rolle nehmen Sie nach der Batterieverordnung ein? Sind Batterien in Ihrem Produkt entfernbar? Je nach Rolle füllt sich die Aufgabenliste rasant weiter: Konformitätsbewertung durchführen, technische Dokumentation erstellen, in Registern melden, Kennzeichnung prüfen, Managementsysteme für die Sorgfaltspflicht vorbereiten, die erweiterte Herstellerverantwortung umsetzen…
Sie fragen sich, welche Pflichten als Hersteller, Importeur oder Händler auf Sie zukommen?
Die Konformitätsbewertung und technische Dokumentation bereiten Ihnen Kopfzerbrechen?
Oder stehen Sie vor der Aufgabe, Ihr Medizinprodukt anzupassen – sei es für die Austauschbarkeit der Gerätebatterie oder gar eine Anpassung im Qualitätsmanagementsystem zur Erfüllung neuer Sorgfaltspflichten?
seleon ist Ihr Partner für diese Herausforderungen.
Mit Erfahrung, Know-how und praxisnahen Lösungen begleiten wir Sie zuverlässig auf dem Weg zu mehr Sicherheit und Compliance.
Bitte beachten Sie, dass alle Angaben und Auflistungen nicht den Anspruch der Vollständigkeit haben, ohne Gewähr sind und der reinen Information dienen.

